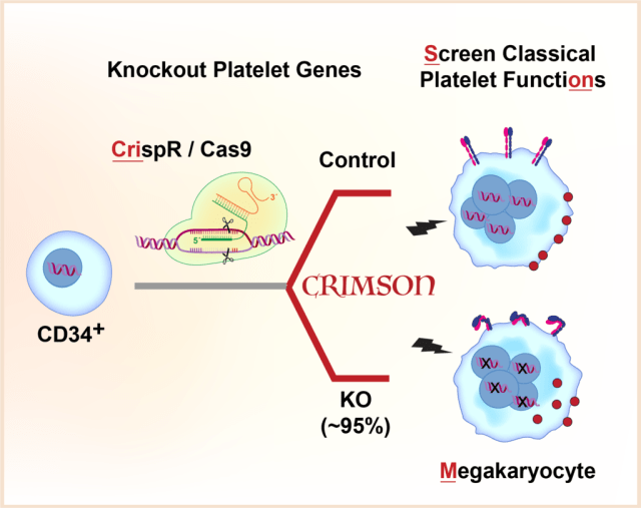
Human platelets are anucleate and cannot be directly modified using traditional genetic approaches. Here we present a rapid, highly efficient approach to screen genes for platelet like functions in primary megakaryocytes called CRIMSON: CRIspr edited Megakaryocytes for rapid Screening of platelet gene function. CRIMSON can be used to test single genes, or multiple genes simultaneously or in parallel.
About Crimson
CRIspr edited Megakaryocytes for rapid screening of platelet gene functions
This website is a resource for the methods used in our Blood Advances paper: https://doi.org/10.1182/bloodadvances.2020004112 Emilie Montenont, Seema Bhatlekar, Shancy Jacob, Yasuhiro Kosaka, Bhanu K. Manne, Olivia Lee, Ivan Parra-Izquierdo, Emilia Tugolukova, Neal D. Tolley, Matthew T. Rondina, Paul F. Bray, Jesse W. Rowley; CRISPR-edited megakaryocytes for rapid screening of platelet gene functions. Blood Adv 2021; 5 (9): 2362–2374.
If you want to make point mutations in megakaryocytes with CRIMSON HD (CRIMSON with Homology Directed Repair), which was presented at ISTH 2022, please check back later, or email the authors.
Human platelets are anucleate and cannot be directly modified using traditional genetic approaches. Here we present a rapid, highly efficient approach to screen genes for platelet like functions in primary megakaryocytes called CRIMSON: CRIspr edited Megakaryocytes for rapid Screening of platelet gene functiON. CRIMSON can be used to test single genes, or multiple genes simultaneously or in parallel.
Why Crimson?
Because platelets are anucleate, human platelet gene function studies depend on mouse models and alternative cell models. As the nucleated precursor to platelets, primary megakaryocytes are the nearest cell to platelets in origin, structure, and function. But a systematic approach to 1. introduce genetic deletions and 2. survey platelet functional responses in megakaryocytes has not been described.
CRIMSON fills the gaps:
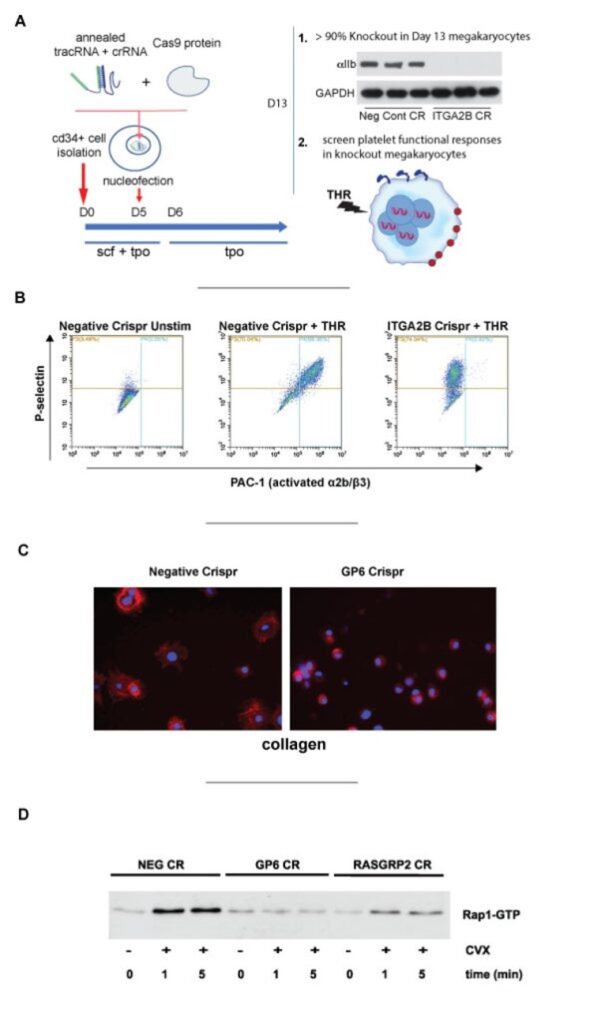
A. Rapid and consistent genetic modifications in CD34+ derived human megakaryocytes without using viruses or selection
- genetic knockout in >90% of functional megakaryocytes, with nearly complete loss of protein for almost any gene using CRISPR/CAS9 nucleofection. No viruses, sorting, or selection needed.
B. Megakaryocytes with platelet gene knockouts model select platelet functional responses
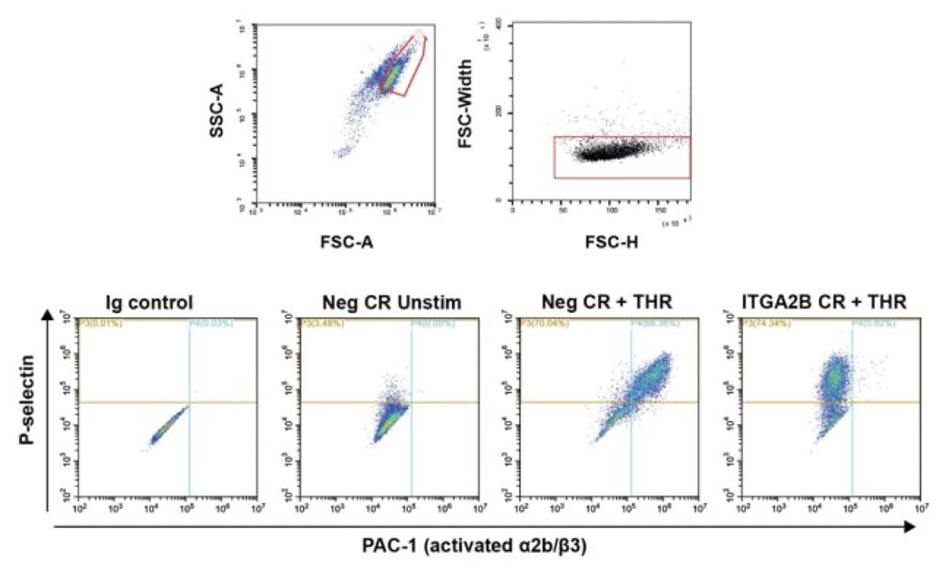
- knockout of ITGA2B abolishes binding of PAC-1 to activated integrin in response without affecting P-selectin translocation.
- knockout of GP6 abolishes megakaryocyte spreading on Collagen.
- knockout of RASGRP2 (CALDAG-GEFI) blunts Rap1b-GTP activation.
Protocol
References and Resources
Emilie Montenont, Seema Bhatlekar, Shancy Jacob, Yasuhiro Kosaka, Bhanu K. Manne, Olivia Lee, Ivan Parra-Izquierdo, Emilia Tugolukova, Neal D. Tolley, Matthew T. Rondina, Paul F. Bray, Jesse W. Rowley; CRISPR-edited megakaryocytes for rapid screening of platelet gene functions. Blood Adv 2021; 5 (9): 2362–2374. doi: https://doi.org/10.1182/bloodadvances.2020004112
The protocol for CRISPR/CAS9 nucleofection was adapted from previous publications:
Bak RO, Dever DP, Porteus MH. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018;13(2):358–376. https://pubmed.ncbi.nlm.nih.gov/29370156/
The following protocol was also used from idtDNA’s website that has since been removed:
Hendel, Ayal; Shapiro, Jenny; Tovin, Ada; Iancu, Ortal; Mizrahi K. Electroporation of primary human CD34+ hematopoietic stem and progenitor cells. Integr. DNA Technol. Inc. 2018;
However the authors have now published a similar article here:
Shapiro J, Iancu O, Jacobi AM, et al. Increasing CRISPR Efficiency and Measuring Its Specificity in HSPCs Using a Clinically Relevant System. Mol Ther Methods Clin Dev. 2020;17:1097-1107. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7251314/
Our first use of the CRISPR method in megakaryocytes to study hematopoiesis was published in:
Bhatlekar S, Manne BK, Basak I, Edelstein LC, Tugolukova E, Stoller ML, Cody MJ, Morley SC, Nagalla S, Weyrich AS, Rowley JW, O’Connell RM, Rondina MT, Campbell RA, Bray PF. miR-125a-5p regulates megakaryocyte proplatelet formation via the actin-bundling protein L-plastin. Blood. 2020 Oct 8;136(15):1760-1772. https://pubmed.ncbi.nlm.nih.gov/32844999/
Some of the megakaryocyte platelet function flow cytometry assays were optimized and expanded based on methods published here:
Basak I, Bhatlekar S, Manne BK, et al. miR-15a-5p regulates expression of multiple proteins in the megakaryocyte GPVI signaling pathway. J Thromb Haemost. 2019;17(3):511-524. https://pubmed.ncbi.nlm.nih.gov/30632265/
idtDNA has many useful guides and protocols for CRISPR design, transfection, and assessing efficiency and off target effects
Materials
All reagents/consumables should be sterile and RNAse/DNAse free
- StemSpan SFEM (Stemcell Technologies)*
- *10/21 update: we recently experienced low cell viability with precipitates forming in several batches of SFEM. We have switched to SFEM II which yields more cells and comparable KO and end phenotypes. Thaw and store the SFEM II as directed by the manufacturer.
- SCF (peprotech) – resuspend to 100 ug/mL in SFEM, aliquot and store at -80c.
- TPO (peprotech) – resuspend to 100 ug/mL in SFEM, aliquot and store at -80c.
- P3 Primary Cell 4D-NucleofectorTM X Kit S https://bioscience.lonza.com/lonza_bs/US/en/Transfection/p/000000000000197190/P3-Primary-Cell-4D-Nucleofector-X-Kit-S
- Sterile RNAse free 10 mM Tris (PH 7.5). We use RNAse free Tris from Ambion. This can be prepared by combining and diluting 1 M Tris PH 8.0 with 1 M Tris PH 7.0 as in https://assets.fishersci.com/TFS-Assets/LSG/manuals/sp_9851.pdf
- Sterile RNAse/DNAse free PBS.
From idtDNA https://www.idtdna.com/pages/products/crispr-genome-editing/alt-r-crispr-cas9-system :
- Alt-R® CRISPR-Cas9 crRNA (used in our publication), Alt-R® CRISPR-Cas9 crRNA XT, or Alt-R® CRISPR-Cas9 sgRNA. See below for design and purchasing.
- Alt-R® CRISPR-Cas9 tracrRNA (to complex with crRNA, don’t need this for the linked sgRNA).
- Alt-R® S.p. Cas9 Nuclease V3.
- Alt-R® Cas9 Electroporation Enhancer.
Equipment and supplies
- Amaxa 4D device. Others have successfully used the Neon system for transfecting CD34+ cells. See idtDNA and Neon websites for using the Neon system.
- Hemacytometer or other method for cell counting.
- Thermal cycler (PCR machine) or heat block with heated lid.
- Incubator set at 37c 5% co2 (note, some recommend low o2 (5%) for MK culture, but this not done in our experiments).
- 1.7 mL eppendorf tubes, RNAse/DNAse free.
- RNAse/DNAse free pipettor and tips.
- 24 well and 6 well tissue culture plates.
- 0.2 mL sterile RNAse/DNAse free PCR tube 8 well strips.
- Sterile tissue culture hood – all steps should be done in sterile RNAse/DNAse free conditions.
For MK phenotyping and platelet function assays
- Thrombin, Sigma Aldrich.
- CRP-XL, CambCol Laboratories.
- Convulxin, ENZO life sciences, ALX-350-100-C050 or Santa Cruz, sc-202554. Note that we have observed significant variation in potency between vials, so always use convulxin from the same vial within an experiment.
- 2-methylthio-ADP (2-MeSADP).
- CD61-FITC, BD biosciences.
- CD41a-APC, BD biosciences, #559777
- CD42a-PE, BD biosciences, #558819
- PAC1-Fitc, BD Biosciences, #340507
- P-sel-PE, BD Biosciences, #555524
- CD63 V450 BD Biosciences, #561984
- Matching isotype controls for antibodies.
- Alex-Fluor 647 labeled Fibrinogen from human plasma (Invitrogen, #F35200).
- 4% paraformaldehyde, (8% PFA from electron microscopy sciences diluted with 2x PBS).
- Collagen, 1 mg/mL (type I, Chrono-log #385).
- Fibrinogen (from human plasma, EMD-Millipore #341576). Reconstitute in sterile water to 10 mg/mL, aliquot and store -80c.
- Alexa Fluor 488 phalloidin, ThermoFisher, #A12379
CRISPR/CAS9 in CD34 + cells
CRISPR design and preparation
- Click here for a Table of crRNA sequences validated with CRIMSON OR design crRNA using idtDNA webtool: https://www.idtdna.com/site/order/designtool/index/CRISPR_PREDESIGN. See note a. below.
- Order 2-3 crRNA (2 nmol) with on-target score > 60%. Be sure to order negative control crRNA.
- Prepare crRNA/tracRNA complexes (not needed for sgRNA):
- Resuspend crRNA to 200 uM (2 nmol in 10 uL) RNAse free 10 mM Tris (PH 7.5)*
- Resuspend tracRNA to 200 uM (5 nmol in 25 uL) RNAse free 10 mM Tris (PH 7.5).
- Mix crRNA 1:1 with tracRNA in RNAse free PCR tube. Mix at least 2.5 uL of each.
- Heat at 95c for 5 min in a thermal cycler with heated lid.
- During step d, resuspend enhancer to 100 uM (2 nmol in 20 uL) in RNAse free 10 mM Tris (PH 7.5), place on ice.
- Remove crRNA/tracRNA from heat and cool to room temperature on bench top.
- Keep at room temperature for 10 min., then place on ice.
- Proceed to CRISPR/CAS9 transfection or store crRNA/tracRNA duplex at -20c.
There are many options for CRISPR design software. We prefer using idtDNA for custom or pre-designed crRNA. We usually check the top 10 designs and aim for crRNA that have >60% on-target score. You will want to test 2-3 non-overlapping crRNA, since not all will work the first time (in our experience at least one of three will, see figure below). Combining two different crRNA often results in even better KO efficiency. We usually choose the crRNA 2 nmol initially. The XT and sgRNA (which doesn’t require tracRNA) versions may work better for some targets (we have tried both), but aren’t usually necessary since we can usually identify a crRNA with sufficiently high efficiency. Depending on the experiment, crRNA with higher off-target scores are preferred. Off targets can be examined through their website to make sure no predicted targets are platelet function genes.
CD34+ cell culture
CD34+ cells are cultured in SFEM/TPO/SCF for 3 days and then in SFEM/TPO until day 13. Alternatively, for a larger cell yield, CD34+ cells can be pre-expanded for 5-6 days ~50 fold and frozen in aliquots for future use using the cocktial IL6, SRI, UM171,Flt3L, TPO, SCF published in https://pubmed.ncbi.nlm.nih.gov/29370156/. Thawed, pre-expanded cells are treated as day 0 cells and cultured using the same protocol below:
- thaw C34+ cord blood cells or isolate fresh using magnetic selection.
- Add 0.5-1×10^6 cells/mL cells to prewarmed SFEM-ST media (cytokines added fresh) in tissue culture plate:
- SFEM + gentamycin*
- 25 ng/mL SCF
- 20 ng/mL TPO
- Incubate at 37c 5% co2.
- passage on day 3 by centrifugation (300xg), replate at 0.5-1×10^6 cells/mL in SFEM-ST media.
- Transfect cells (see next section) between day 1 and day 6 (we prefer day 5) and replate in SFEM-ST media.
- passage on day 6 and 9 or 10 as in step 4, but use SFEM-T media. Starting on day 6, keep cells in larger wells (i.e. 1 mL (0.5-1×10^6/mL) in a 12 well plate, 2-3 mL in a 6 well plate) to maximize the surface area exposed to air.
- SFEM + gentamycin*
- 50 ng/mL TPO**
*Gentamycin is optional but recommended to prevent bacterial growth. Gentamycin is known to affect mitochondria in other mammalian cells. 10/21 update: switched to using Pen/Strep as the antibiotic of choice with no noticeable differences.**use 100 ng/mL TPO day 6 on, and keep cells at 0.5×10^6/mL for enhanced proplatelet formation.
CRISPR/CAS9 transfection
On the day of electroporation:
- Pre-warm SFEM-ST media.
- Add 425 uL (for 1×10^6 cells) of media per transfection to each well of a 24 well plate. To another well, add extra 100 uL per transfection. place in incubator to warm.
- Place Amaxa solution P3 (supplemented) at room temperature.
Prepare cells:
- Resuspend cells in the wells by gently pipetting.
- Transfer cells to a 15 mL sterile tube.
- Rinse the culture dish surface with an additional 1 mL of prewarmed culture medium and add this volume to the tube.
- Mix cells gently, count and assess viability (>90%) by hemacytometer or automated counter
- Transfer the number of cells needed into a new 15 mL sterile tube. Between 2.5e5 and 4e6 cells per transfection. We prefer 1e6 cells/transfection.
- Place the cells in incubator with cap loose while preparing rest of experiment.
Prepare CRISPR/CAS9 RNP complexes
If not already done, complex crRNA with tracRNA as described in step 3 of “CRISPR design and preparation above“.
RNP formation:
RNP formation should be done immediately before transfection, while cells are simultaneously being prepared below, and the time between preparation and transfection should be standardized.
- For each transfection, mix the following in a 0.2 mL PCR tube (we prefer 8 tube strips), gently swirling the pipet tip while pipetting. Sufficient for 2 transfections, scale up as necessary.
- PBS (sterile/RNAse free) – 2.1 uL
- crRNA/tracRNA duplex – 1.2 uL (120 pmol)
- Alt-R spCas9 nuclease V3 (provided as 62 uM stock) – 1.7 uL (105 pmol)
- Heat RNP complexes at 37c in thermal cycler for 4 min.
- While RNP is forming, immediately centrifuge cells at 300xg, 5 min, room temp.
- Remove RNP complexes to room temperature for 10 min prior to nucleofection. Prolonged incubation may reduce efficiency.
- During step 4, immediately resuspend cells in 1 mL PBS, transfer to a sterile microtube, rinse original tube with 500 uL PBS and add rinse to rest of sample.
- Centrifuge cells again at 300xg, 5 min, room temp.
- Completely remove supernatant.
- Resuspend cells in 20 uL per transfection of room temp, supplemented, solution P3 (i.e. for 4 transfections at 1e6 cells/transfection, add 80 uL to 4e6 cells).
- Transfer 20 uL of the cell suspension into individual sterile PCR tubes.
- Proceed immediately to nucleofection.
Amaxa nucleofection:
- Working quickly (very important – efficiency will improve with practice), add RNA complexes and enhancer to cells in PCR tubes as follows:
- Electroporation Enhancer – 1 uL (3.85 uM) (note, we now add this to the bottom of the PCR tube in advance while cells are centrifuging.)
- Cells – 20 uL
- RNP complex from “RNP formation” step – 2.5 uL (final is 4.6 uM crRNA/tracRNA, 4 uM cas9, may need to titrate for different loci (0.5 – 4 uM))*
- Avoiding bubbles, pipet mixture up and down 2 times (consider using multichannel for multiple transfections) and transfer 25 uL to the 16 well electroporation strip. Note: the cuvette can only be loaded onto the nucleofector in 1 direction, so check you are loading samples accordingly.
- On the Amaxa device select the volume/wells to be transfected, program DZ100
- Select ok to load strip
- Place nucleofector strip into the 4D device
- Select start
- Immediately retrieve media from the incubator and add 75 uL prewarmed media (from the extra well on the plate) to each well. Let sit for 3-5 min at room temp.
- Gently transfer the entire volume to the 425 uL SFEM-ST media in the 24 well plate
- Incubate 24 hours. Usually we transfect on day 5 and passage on day 6 to fresh SFEM-T media and continue culture as described in the section CD34+ Cell Culture. Visually inspect cells on day 8 by light microscope – see example below:
Assessing cutting efficiency
Light microscope image (20x objective) of a well of CRISPR/CAS9 transfected CD34+ cells 3 days after transfection (day 8 of culture)
*For transfecting multiple crRNA at the same time, add 2.5 uL of each RNP complex.
Assessing cutting efficiency:
Cutting efficiency can be assessed as soon as 3 days after transfection. Since MKs become multinucleate, we usually assess cutting on day 13 cells.
- Pellet 2×10^5 cells from KO and control
- Isolate DNA (i.e. Qiagen genomic DNA extraction kit)
- Design primers to amplify that flank ~200 basepairs on one side and 400 base pairs on the other side of the target site
- Amplify cut region via PCR
- Clean up PCR and perform Sanger sequencing
- Plug KO and control sanger sequencing files into TIDE analysis for total efficiency score: https://tide.nki.nl/ or Synthego ICE for KO score: https://www.synthego.com/products/bioinformatics/crispr-analysis
Note that these software seem to sometimes underestimate by 10-20% the actual % of knock out we observe by protein analysis.
Megakaryocyte phenotypic and platelet function assays
Assays performed on day 13 Megakaryocytes. Count cells and resuspend in SFEM without growth factors at 1×10^6/mL . All assays should be done in comparison to a non-targeting crRNA control and if desired a targeting control (i.e. B2M).
Proplatelet formation
- Coat 8 well borosilicate chamber slide with 100 ug/mL fibrinogen diluted in PBS for 30 min at 37c.
- Remove fibrinogen, block with 2.5% human serum albumin for 20 minutes.
- Wash 3x PBS.
- Add 2.5×10^5 cells in 250 uL SFEM-T per well.
- Incubate overnight.
- Add 100 uL 4% PFA for 20 minutes at RT.
- Fix with 2% paraformaldehyde.
- Wash 3x with PBS.
- Permeabilize cells with 0.1% triton x 100 for 5 minutes.
- Wash 3x with PBS.
- Add phalloidin 488 and nuclear stain for 15 minutes at RT.
- Wash 3x with PBS, keep in PBS for imaging.
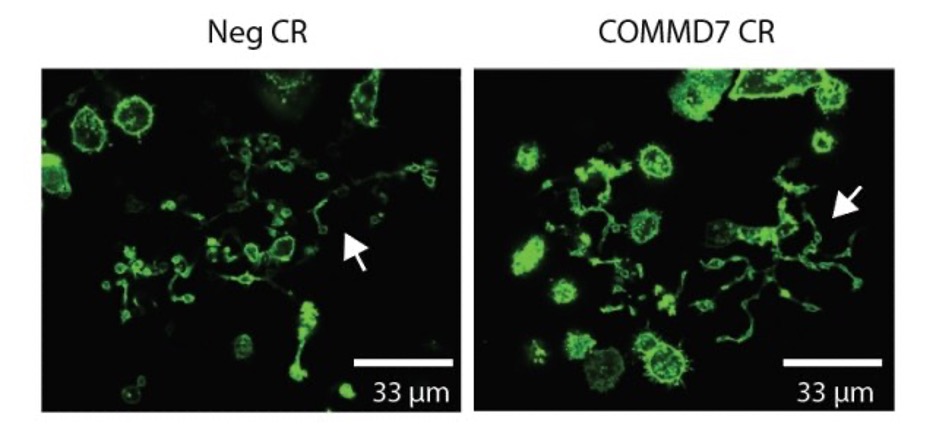
- While blinded to treatment, image cells on fluorescent microscope, take ratio of # proplatelet forming vs total cells. Examples of proplatelet forming megakaryocytes are marked with white arrows in the images below:
Spreading
- Coat duplicate wells of 96 well plates with collagen (coated the night before) or fibrinogen.
- Dilute collagen 1:10 in HBSS. 40 uL to each well, place at 4c overnight.
- Thaw reconstituted fibrinogen at 37c. Dilute 1:10,000 in HBSS, 40 uL each well place for 30 minutes at 37c.
- Wash wells 3 times with PBS.
- Add 2.5×10^4 megakaryocytes to wells in 100 uL media.
- Incubate 1 hour at 37c.
- Gently remove unbound cells.
- Wash gently with PBS.
- Fix with 2% paraformaldehyde.
- Process cells for imaging as in steps 8-12 of proplatelet assay.
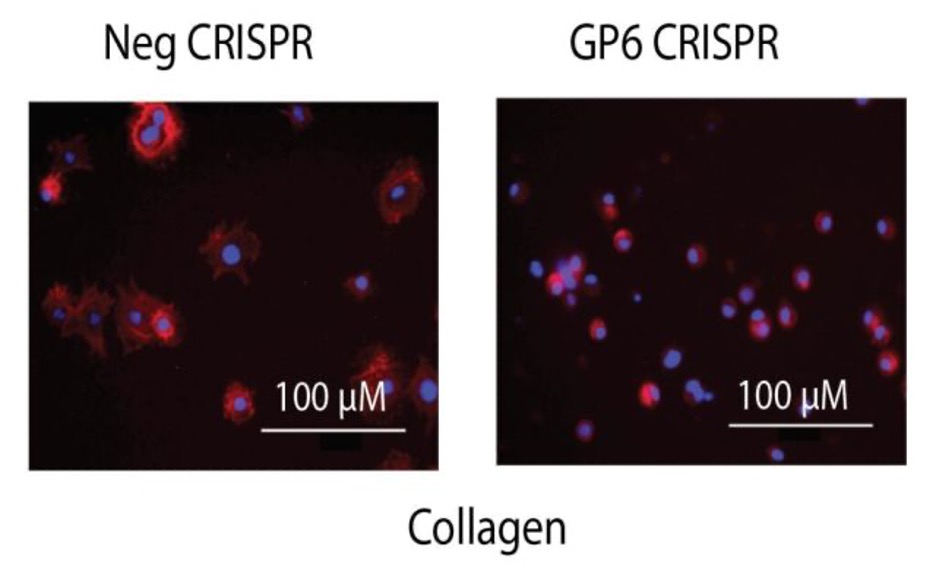
- While blinded to treatment, image cells on fluorescent microscope, take ratio of # spread vs round cells. In the example below most of the negative CRISPR treated cells are spread (egg on a pan) on colagen, whereas most GP6 KO cells are round.
Flow cytometry phenotyping and assays of activation
We do activation assays in a round bottom polypropylene 96 well plate. Include an unstimulated activated and unactivated matching isotype control well for at least one of the conditions.
1. Prealiquot 50 uL of phenotyping, activation, or fibrinogen binding antibody mix into individual wells. Use SFEM without cytokines:
Phenotyping mix
- 1 uL CD41-APC
- 2 uL CD42a-PE
- 1 uL CD61-FITC
- 46 uL SFEM
Activation mix
- 6 uL cd62-PE (granule marker)
- 10 uL PAC1-FITC (integrin activation)
- 5 uL CD63-V450 (dense granules/lysosomes, optional)
- 1 uL CD41-APC (MK marker)
- 28 uL SFEM
Fibrinogen binding mix
- 2 uL CD42a-PE
- 1 uL Fibrinogen-647
- 47 uL SFEM
2. Add vehicle (for unstimulated) or agonist, such as one of the following, to activation wells:
- 1 U/mL thrombin*
- 1.25 ug/mL convulxin
- 2 ug/mL CRP
- 200 nM 2-MesSADP
*Thrombin can’t be used in the fibrinogen binding assay without adding inhibitors to prevent clot formation
3. Immediately add 50 uL of cells (5×10^5) and pipet to mix (for many conditions use a multichannel pipetor
4. Incubate at 37c for 15 minutes
5. Fix with 100 uL 4% PFA (2% final)
6. Analyze by flow cytometry
Gate on higher FSC, lower SSC population as described by https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6886406/ . Further gate on singlets using FSC-H and FSC-W. Set phenotype marker (CD41, CD61,CD42a) or activation gates (PAC-1, P-selectin, or CD63) based on isotype controls. Record % positive cells and gMFI of the gated cells. Gating examples shown below for negative control vs ITGA2B KO cells.
CRIMSON validated CRISPR sequences
| Gene | idtDNA id | Sequence | Cutting efficiency | % KO (protein) |
| B2M | AA + AB | CGTGAGTAAACCTGAATCTT + AAGTCAACTTCAATGTCGGA | ND | 95% (2 crRNA together), western |
| ITGA2B | AF | GGACAAGCGTTACTGTGAAG | 75%, synthego | 99%, western |
| GP6 | AL | CATACCGAGTCTGACACTGT | 58%, TIDE | 85%, western |
| RASGRP2 | AJ | CCTGGACAAGGGCTGCACGG | 83%, TIDE | 95%, western |
| COMMD7 | AA + AC | ACCTATGGCCCATCGAGCAA + ACCACCAATCAGATCAGTCT | ND | 96% (2 crRNA together), western |